Fabry-sjukdomen
Fabrys sjukdom är en lysosomal lagringssjukdom som ärvs på ett X-bundet recessivt sätt, orsakad av en mutation i GLA-genen. Som ett resultat av denna mutation uppstår en brist på enzymet alfa-galaktosidas A, som ansvarar för att bryta ner fettämnen. Detta leder till ansamling av globotriaosylceramid i olika organ.
Fabry-sjukdomen tillhör gruppen ultrasällsynta mukopolysackaridoser. Tillståndet kan orsaka svåra episoder av outhärdlig, brännande smärta i de distala delarna av extremiteterna, som strålar ut till olika delar av kroppen.
Typiska symtom inkluderar också termoreglering och svettningsstörningar, även i stressiga situationer. Patienter dör främst av för tidig hjärt- eller hjärnskada före 50 års ålder. På grund av symtomens ospecifika natur kan det ta flera år att få en korrekt diagnos.
Orsaker till Fabrys sjukdom
Fabrys sjukdom klassificeras som en sällsynt lysosomal lagringsstörning. Den orsakas av en mutation i GLA-genen, vilket leder till antingen en fullständig eller signifikant minskning av aktiviteten hos alfa-galaktosidas A-enzym.
Följaktligen ackumuleras globotriaosylceramid (GL-3) i cellerna i olika vävnader och organ, eftersom enzymbristen förhindrar dess nedbrytning. Ansamlingen av GL-3 stör de drabbade organens funktioner och orsakar komplikationer.
Denna sällsynta sjukdom är X-bunden, vilket innebär att den förekommer oftare hos män. Dock kan heterozygota kvinnor också uppvisa symtom, med varierande svårighetsgrad. Hos kvinnor visar sig sjukdomens kliniska manifestation vanligtvis flera år senare än hos män.
Diagnos av Fabrys sjukdom
För att bekräfta diagnosen Fabrys sjukdom hos män mäts aktiviteten av alfa-galaktosidas A i plasma, leukocyter eller fibroblaster. Om enzymbrist upptäcks utförs genetisk testning. Hos kvinnor, eftersom alfa-galaktosidas A-aktivitet kan ligga inom normala intervall, krävs genetisk testning för att identifiera bärare av den defekta gen som orsakar Fabrys sjukdom.
Symtom på Fabrys sjukdom
Den visar sig i två fenotyper:
- Klassisk fenotyp – Vanligare hos män, kännetecknad av frånvaro eller minimal enzymaktivitet, med symtom som uppträder i barndom eller tonåren.
- Icke-klassisk fenotyp – Symtomen uppstår senare i livet på grund av en lägre grad av alfa-galaktosidas A-brist.
Symtom som uppstår under tonåren inkluderar:
- Intolerans mot höga temperaturer
- Nedsatt svettning
- Hypertermi
- Parestesi (intensiv brännande smärta i de distala extremiteterna)
- Buksmärta, förstoppning, diarré, illamående och kräkningar
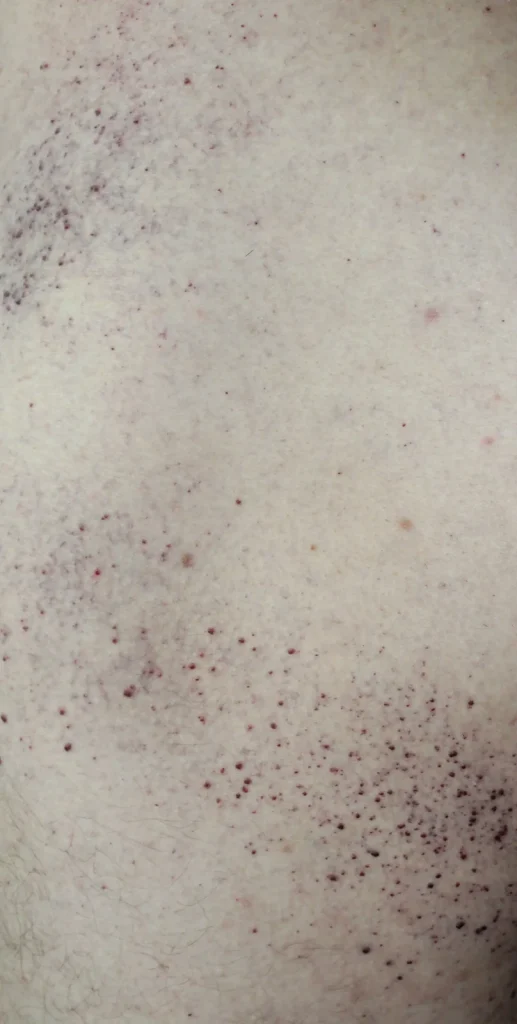
- Hudskador på lår, skinka och nedre delen av buken i form av rödlila utslag (angiokeratom)
- Hornhinnemoln, grå starr och progressiv hörselnedsättning
I vuxen ålder inkluderar symtomen:
- Strukturella och funktionella kardiovaskulära avvikelser (vänster ventrikelhypertrofi, arytmier och ledningsstörningar)
- Njurskada, som initialt visade sig som proteinuri och mikroalbuminuri
- Kronisk trötthet, tinnitus och yrsel
Dessutom är beteendeförändringar som ångest, social isolering, depression och skuldkänslor vanliga. Många av dessa symtom uppträder före diagnos, eftersom Fabry-patienter, liksom de med andra sällsynta sjukdomar, ofta upplever långa diagnostiska förseningar som påverkar både fysisk och psykisk hälsa. Studier visar att livskvaliteten hos Fabry-patienter är betydligt lägre jämfört med den allmänna befolkningen.
Prognos
De vanligaste dödsorsakerna hos Fabry-patienter är stroke och hjärtinfarkt. Jämfört med den allmänna befolkningen har män med Fabrys sjukdom en förväntad livslängd som minskats med 15–20 år, medan kvinnor lever ungefär 6–10 år kortare.
Behandlingsalternativ
Standardbehandlingen för Fabrys sjukdom är enzymersättningsterapi (ERT) med agalsidas, alfa eller beta. Med tanke på sjukdomens multisystemiska inblandning är stödjande terapier också nödvändiga för att hantera komplikationer och förhindra ytterligare hälsoförsämring.
ERT kan bromsa sjukdomsutvecklingen och göra det möjligt för patienter att upprätthålla familje-, arbets- och sociala aktiviteter.
Dessutom har migalastat godkänts för behandling av Fabrys sjukdom. Till skillnad från intravenös ERT är migalastat en oral medicin. Dock kan endast patienter med specifika mutationer som svarar på detta läkemedel få det – ungefär 35–50 % av alla Fabry-patienter.
Stödjande terapier för Fabrys sjukdom inkluderar:
- Smärtbehandling, främst genom farmakoterapi
- Neuroprotektiva åtgärder (angiotensinhämmare och angiotensinreceptorblockerare)
- Antiarytmiska läkemedel
- Pacemaker eller implanterbar kardioverter-defibrillator (ICD) för svår kardiovaskulärpåverkan
- Dialys eller njurtransplantation vid avancerad njursvikt
Behandlingen av Fabrys sjukdom är livslång, och tidig intervention ökar risken för att fördröja eller förebygga allvarliga komplikationer.
Förekomst
Globalt uppskattas dess förekomst till 1 på 40 000 till 1 på 170 000 individer. Dock kan dessa siffror underskattas på grund av komplexiteten i Fabrys sjukdoms symtom och utmaningar med differentialdiagnos.