Fabryziekte
De ziekte van Fabry is een lysosomale opslagstoornis die op een X-gebonden recessieve manier wordt overgeërfd, veroorzaakt door een mutatie in het GLA-gen. Als gevolg van deze mutatie is er een tekort aan het enzym alfa-galactosidase A, dat verantwoordelijk is voor het afbreken van vetstoffen. Dit leidt tot de ophoping van globotriaosylceramide in verschillende organen.
De Fabry-ziekte behoort tot de groep van ultrazeldzame mucopolysaccharidoses. De aandoening kan ernstige episodes van ondraaglijke, brandende pijn veroorzaken in de distale delen van de ledematen, die zich uitstralen naar verschillende delen van het lichaam.
Kenmerkende symptomen zijn ook thermoregulatie en zweetproblemen, zelfs in stressvolle situaties. Patiënten bezwijken voornamelijk aan voortijdige hart- of hersenschade vóór de leeftijd van 50 jaar. Door de niet-specifieke aard van de symptomen kan het krijgen van een juiste diagnose meerdere jaren duren.
Oorzaken van Fabry-ziekte
De ziekte van Fabry wordt geclassificeerd als een zeldzame lysosomale opslagstoornis. Het wordt veroorzaakt door een mutatie in het GLA-gen, wat leidt tot een volledige of significante vermindering van de activiteit van het enzym alfa-galactosidase A.
Daardoor hoopt globotriaosylceramide (GL-3) zich op in de cellen van verschillende weefsels en organen, omdat het enzymtekort de afbraak ervan verhindert. De ophoping van GL-3 verstoort de functies van aangetaste organen en veroorzaakt complicaties.
Deze zeldzame ziekte is X-gebonden, wat betekent dat het vaker voorkomt bij mannen. Echter, heterozygote vrouwen kunnen ook symptomen vertonen, met verschillende ernstgradaties. Bij vrouwen verschijnt de klinische manifestatie van de ziekte meestal enkele jaren later dan bij mannen.
Diagnose van de ziekte van Fabry
Om een diagnose van de ziekte van Fabry bij mannen te bevestigen, wordt de activiteit van alfa-galactosidase A gemeten in plasma, leukocyten of fibroblasten. Als enzymtekort wordt vastgesteld, wordt genetisch onderzoek uitgevoerd. Bij vrouwen, aangezien de activiteit van alfa-galactosidase A binnen normale waarden kan blijven, is genetisch onderzoek noodzakelijk om dragers van het defecte gen dat verantwoordelijk is voor de ziekte van Fabry te identificeren.
Symptomen van de ziekte van Fabry
Het komt voor in twee fenotypen:
- Klassiek fenotype – Vaker voorkomend bij mannen, gekenmerkt door afwezige of minimale enzymactiviteit, met symptomen die optreden in de kindertijd of adolescentie.
- Niet-klassiek fenotype – Symptomen verschijnen later in het leven door een minder ernstige mate van alfa-galactosidase A-tekort.
Symptomen die zich in de adolescentie voordoen, zijn onder andere:
- Intolerantie voor hoge temperaturen
- Verminderd zweten
- Hyperthermie
- Parestesie (intense brandende pijn in de distale ledematen)
- Buikpijn, constipatie, diarree, misselijkheid en braken
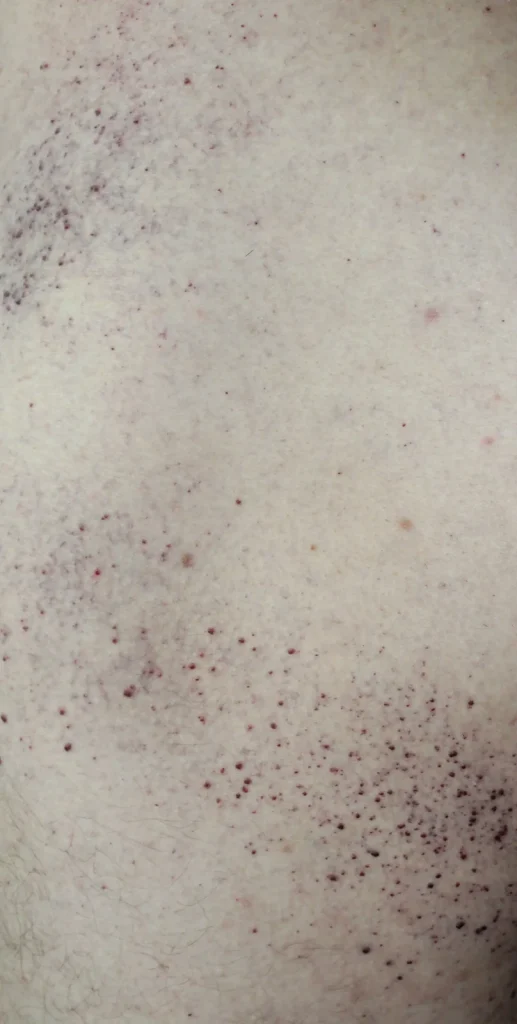
- Huidlaesies op de dijen, billen en onderbuik in de vorm van rood-paarse uitslag (angiokeratoom)
- Hoornvlechtvertroebeling, staar en progressief gehoorverlies
Op volwassen leeftijd zijn symptomen onder andere:
- Structurele en functionele cardiovasculaire afwijkingen (linkerventrikelhypertrofie, aritmieën en geleidingsstoornissen)
- Nierschade, aanvankelijk gepresenteerd als proteïnurie en microalbuminurie
- Chronische vermoeidheid, tinnitus en duizeligheid
Daarnaast komen gedragsveranderingen zoals angst, sociale isolatie, depressie en schuldgevoelens vaak voor. Veel van deze symptomen verschijnen al vóór de diagnose, aangezien Fabry-patiënten, net als die met andere zeldzame ziekten, vaak lange diagnostische vertragingen ervaren, wat zowel het fysieke als het mentale welzijn beïnvloedt. Uit onderzoek blijkt dat de kwaliteit van leven bij Fabry-patiënten aanzienlijk lager is dan in de algemene bevolking.
Prognose
De meest voorkomende doodsoorzaken bij Fabry-patiënten zijn een beroerte en een hartaanval. In vergelijking met de algemene bevolking hebben mannen met de ziekte van Fabry een levensverwachting die met 15-20 jaar is verkort, terwijl vrouwen ongeveer 6-10 jaar minder leven.
Behandelingsopties
De standaardbehandeling voor de ziekte van Fabry is enzymvervangingstherapie (ERT) met agalsidase, alfa of bèta. Gezien de multisystemische betrokkenheid van de ziekte zijn ondersteunende therapieën ook noodzakelijk om complicaties te beheersen en verdere gezondheidsachteruitgang te voorkomen.
ERT kan de ziekteprogressie vertragen en patiënten in staat stellen familie-, werk- en sociale activiteiten te onderhouden.
Daarnaast is migalastat goedgekeurd voor de behandeling van de ziekte van Fabry. In tegenstelling tot intraveneuze ERT is migalastat een orale medicatie. Echter, alleen patiënten met specifieke mutaties die responsief zijn op dit medicijn kunnen het ontvangen—ongeveer 35-50% van alle Fabry-patiënten.
Ondersteunende therapieën voor de ziekte van Fabry zijn onder andere:
- Pijnbestrijding, voornamelijk via farmacotherapie
- Neuroprotectieve maatregelen (angiotensine remmers en angiotensine receptorblokkers)
- Anti-aritmiemedicatie
- Pacemaker of implanteerbare cardioverter-defibrillator (ICD) plaatsing voor ernstige cardiovasculaire betrokkenheid
- Dialyse of niertransplantatie bij gevorderde nierstoornis
De behandeling van de ziekte van Fabry duurt het leven lang en vroege interventie vergroot de kans op het uitstellen of voorkomen van ernstige complicaties.
Prevalentie
Wereldwijd wordt de prevalentie geschat op 1 op 40.000 tot 1 op de 170.000 individuen. Door de complexiteit van de symptomen van Fabry-ziekte en de uitdagingen bij differentiële diagnose kunnen deze cijfers echter worden onderschat.