Malattia di Fabry
La malattia di Fabry è un disturbo di accumulo lisosomale ereditato in modo recessivo legato all’X X, causato da una mutazione nel gene GLA. A causa di questa mutazione, vi è una carenza dell’enzima alfa-galattosidasi A, responsabile della degradazione delle sostanze grasse. Questo porta all’accumulo di globotriaosilceramide in vari organi.
La malattia di Fabry appartiene al gruppo delle mucopolisaccaridosi ultra-rare. La condizione può causare episodi gravi di dolore lancinante e bruciante nelle parti distali degli arti, che si irradiano a diverse parti del corpo.
I sintomi caratteristici includono anche la termoregolazione e i disturbi della sudorazione, anche in situazioni di stress. I pazienti muoiono principalmente a causa di danni prematuri al cuore o cerebrali prima dei 50 anni. A causa della natura aspecifica dei sintomi, ottenere una diagnosi corretta può richiedere diversi anni.
Cause della malattia di Fabry
La malattia di Fabry è classificata come un raro disturbo della conservazione lisosomiale. È causata da una mutazione nel gene GLA, che porta a una riduzione completa o significativa dell’attività dell’enzima alfa-galattidosasi A.
Di conseguenza, la globotriaosilceramide (GL-3) si accumula nelle cellule di vari tessuti e organi, poiché la carenza enzimatica ne impedisce la decomposizione. L’accumulo di GL-3 altera le funzioni degli organi colpiti e causa complicazioni.
Questa rara malattia è legata all’X X, il che significa che si manifesta più frequentemente nei maschi. Tuttavia, anche le femmine eterozigoti possono manifestare sintomi, con diversi gradi di gravità. Nelle donne, la manifestazione clinica della malattia appare generalmente diversi anni dopo rispetto agli uomini.
Diagnosi della malattia di Fabry
Per confermare la diagnosi della malattia di Fabry negli uomini, l’attività dell’alfa-galattidosasi A viene misurata in plasma, leucociti o fibroblasti. Se viene rilevata una carenza enzimatica, vengono eseguiti test genetici. Nelle donne, poiché l’attività dell’alfa-galattidosidasi A può rimanere entro i valori normali, sono necessari i test genetici per identificare i portatori del gene difettoso responsabile della malattia di Fabry.
Sintomi della malattia di Fabry
Si presenta in due fenotipi:
- Fenotipo classico – Più comune negli uomini, caratterizzato da un’attività enzimatica assente o minima, con sintomi che si manifestano nell’infanzia o nell’adolescenza.
- Fenotipo non classico – I sintomi compaiono più tardi nella vita a causa di un minor grado di carenza di alfa-galattidosidasi A.
I sintomi che compaiono durante l’adolescenza includono:
- Intolleranza alle alte temperature
- Sudore compromesso
- Ipertermia
- Parestesia (dolore intenso e bruciante agli arti distali)
- Dolore addominale, stitichezza, diarrea, nausea e vomito
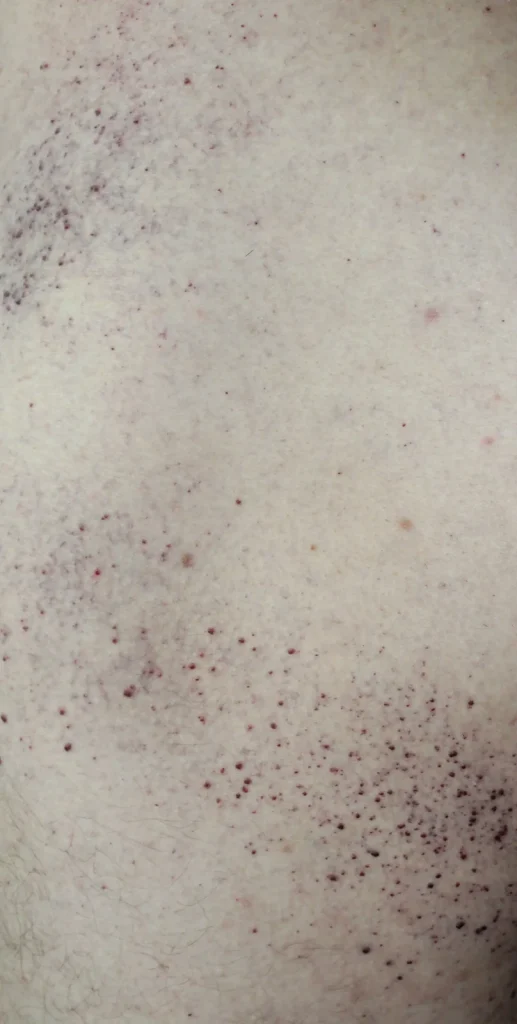
- Lesioni cutanee sulle cosce, glutei e basso addome sotto forma di eruzioni cutanee rosso-viola (angiokeratoma)
- Offuscamento corneale, cataratta e perdita uditiva progressiva
In età adulta, i sintomi includono:
- Anomalie strutturali e funzionali cardiovascolari (ipertrofia ventricolare sinistra, aritmie e disturbi della conduzione)
- Danni renali, inizialmente presentati come proteinuria e microalbuminuria
- Affaticamento cronico, acufene e vertigini
Inoltre, cambiamenti comportamentali come ansia, isolamento sociale, depressione e sensi di colpa sono comuni. Molti di questi sintomi compaiono prima della diagnosi, poiché i pazienti con Fabry, come quelli con altre malattie rare, spesso sperimentano lunghi ritardi diagnostici, che influenzano sia il benessere fisico che mentale. Gli studi mostrano che la qualità della vita nei pazienti con Fabry è significativamente inferiore rispetto alla popolazione generale.
Prognosi
Le cause di morte più comuni nei pazienti con Fabry sono ictus e infarto. Rispetto alla popolazione generale, gli uomini con la malattia di Fabry hanno un’aspettativa di vita ridotta di 15-20 anni, mentre le donne vivono circa 6-10 anni in meno.
Opzioni di trattamento
Il trattamento standard per la malattia di Fabry è la terapia sostitutiva enzimatica (ERT) con agalsidase alfa o beta. Dato il coinvolgimento multisistemico della malattia, sono inoltre necessarie terapie di supporto per gestire le complicazioni e prevenire un ulteriore deterioramento della salute.
L’ERT può rallentare la progressione della malattia e permettere ai pazienti di mantenere la famiglia, il lavoro e le attività sociali.
Inoltre, il migalastat è stato approvato per il trattamento della malattia di Fabry. A differenza dell’ERT endovenosa, il migalastat è un farmaco orale. Tuttavia, solo i pazienti con specifiche mutazioni reattive a questo farmaco possono riceverlo—circa il 35-50% di tutti i pazienti con Fabry.
Le terapie di supporto per la malattia di Fabry includono:
- Gestione del dolore, principalmente attraverso la farmacoterapia
- Misure neuroprotettive (inibitori dell’angiotensina e bloccanti dei recettori dell’angiotensina)
- Farmaci antiaritmici
- Posizionamento di pacemaker o cardioverter-defibrillatore (ICD) impiantabile per un coinvolgimento cardiovascolare grave
- Dialisi o trapianto renale nei casi di deficit renale avanzato
Il trattamento della malattia di Fabry è duraturo e l’intervento precoce aumenta le probabilità di ritardare o prevenire complicazioni gravi.
Prevalenza
A livello globale, la sua prevalenza è stimata tra 1 e 1 su 170.000 individui. Tuttavia, a causa della complessità dei sintomi della malattia di Fabry e delle difficoltà nella diagnosi differenziale, queste cifre potrebbero essere sottovalutate.