Maladie de Fabry
La maladie de Fabry est un trouble lysosomal de stockage hérité de manière récessive liée à l’X et récessif, causé par une mutation du gène GLA. À la suite de cette mutation, il y a une carence de l’enzyme alpha-galactosidase A, responsable de la dégradation des substances grasses. Cela conduit à l’accumulation de globotriaosylcéramide dans divers organes.
La maladie de Fabry appartient au groupe des mucopolysaccharidoses ultra-rares. Cette affection peut provoquer des épisodes sévères de douleurs atroces et brûlantes dans les parties distales des membres, qui irradiant vers différentes parties du corps.
Les symptômes caractéristiques incluent également la thermorégulation et les troubles de la transpiration, même dans des situations stressantes. Les patients succombent principalement à des lésions cardiaques ou cérébrales prématurées avant l’âge de 50 ans. En raison de la nature non spécifique des symptômes, obtenir un diagnostic approprié peut prendre plusieurs années.
Causes de la maladie de Fabry
La maladie de Fabry est classée comme un trouble rare de stockage lysosomal. Elle est causée par une mutation du gène GLA, entraînant soit une réduction complète, soit significative de l’activité de l’enzyme alpha-galactosidase A.
Par conséquent, la globotriaosylcéramide (GL-3) s’accumule dans les cellules de divers tissus et organes, car cette carence enzymatique empêche sa dégradation. L’accumulation de GL-3 perturbe les fonctions des organes affectés et provoque des complications.
Cette maladie rare est liée à l’X, ce qui signifie qu’elle survient plus fréquemment chez les hommes. Cependant, les femelles hétérozygotes peuvent également présenter des symptômes, avec des degrés de gravité variables. Chez les femmes, la manifestation clinique de la maladie apparaît généralement plusieurs années plus tard que chez les hommes.
Diagnostic de la maladie de Fabry
Pour confirmer un diagnostic de la maladie de Fabry chez l’homme, l’activité de l’alpha-galactosidase A est mesurée dans le plasma, les leucocytes ou les fibroblastes. Si une carence enzymatique est détectée, des tests génétiques sont réalisés. Chez les femmes, puisque l’activité de l’alpha-galactosidase A peut rester dans des plages normales, un test génétique est nécessaire pour identifier les porteurs du gène défectueux responsable de la maladie de Fabry.
Symptômes de la maladie de Fabry
Elle se manifeste sous deux phénotypes :
- Phénotype classique – Plus fréquent chez les hommes, caractérisé par une activité enzymatique absente ou minimale, avec des symptômes apparaissant durant l’enfance ou l’adolescence.
- Phénotype non classique – Les symptômes apparaissent plus tard dans la vie en raison d’un degré moindre de carence en alpha-galactosidase A.
Les symptômes apparaissant à l’adolescence incluent :
- Intolérance aux hautes températures
- Sudation altérée
- Hyperthermie
- Paresthésie (douleur brûlante intense dans les membres distals)
- Douleurs abdominales, constipation, diarrhée, nausées et vomissements
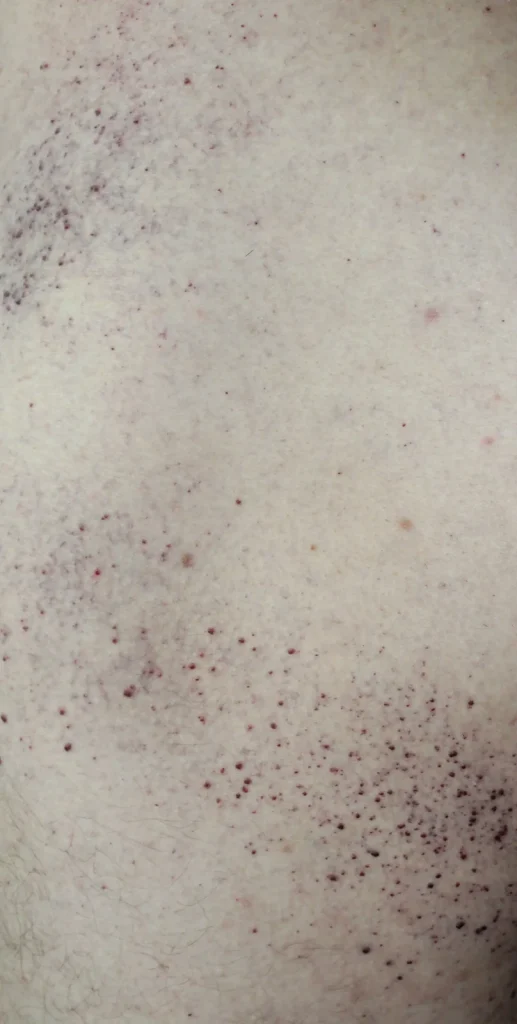
- Lésions cutanées sur les cuisses, les fesses et le bas de l’abdomen sous forme d’éruptions rouge-pourpres (angiocératome)
- Opacité cornéenne, cataractes et perte auditive progressive
À l’âge adulte, les symptômes incluent :
- Anomalies cardiovasculaires structurelles et fonctionnelles (hypertrophie du ventricule gauche, arythmies et troubles de la conduction)
- Lésions rénales, initialement présentes par protéinurie et microalbuminurie
- Fatigue chronique, acouphènes et vertiges
De plus, des changements comportementaux tels que l’anxiété, l’isolement social, la dépression et les sentiments de culpabilité sont fréquents. Beaucoup de ces symptômes apparaissent avant le diagnostic, car les patients atteints de Fabry, comme ceux atteints d’autres maladies rares, rencontrent souvent de longs retards diagnostiques, impactant à la fois le bien-être physique et mental. Des études montrent que la qualité de vie des patients de Fabry est significativement inférieure à celle de la population générale.
Pronostic
Les causes de décès les plus fréquentes chez les patients atteints de Fabry sont l’AVC et la crise cardiaque. Comparés à la population générale, les hommes atteints de la maladie de Fabry ont une espérance de vie réduite de 15 à 20 ans, tandis que les femmes vivent environ 6 à 10 ans de moins.
Options de traitement
Le traitement standard de la maladie de Fabry est la thérapie de remplacement enzymatique (TRE) avec de l’aglasidase alfa ou bêta. Compte tenu de l’implication multisystémique de la maladie, des thérapies de soutien sont également nécessaires pour gérer les complications et prévenir une aggravation de la santé.
La TRE peut ralentir la progression de la maladie et permettre aux patients de maintenir leurs activités familiales, professionnelles et sociales.
De plus, le migalastat a été approuvé pour traiter la maladie de Fabry. Contrairement à la TRE intraveineuse, le migalastat est un médicament oral. Cependant, seuls les patients présentant des mutations spécifiques réactives à ce médicament peuvent le recevoir — environ 35 à 50 % de tous les patients atteints de Fabry.
Les thérapies de soutien pour la maladie de Fabry incluent :
- Gestion de la douleur, principalement par pharmacothérapie
- Mesures neuroprotectrices (inhibiteurs de l’angiotensine et bloqueurs des récepteurs de l’angiotensie)
- Médicaments antiarythmiques
- Placement d’un stimulateur cardiaque ou d’un défibrillateur cardiovertisseur implantable (DCI) pour une atteinte cardiovasculaire sévère
- Dialyse ou greffe rénale dans les cas d’insuffisance rénale avancée
Le traitement de la maladie de Fabry est à vie, et une intervention précoce augmente les chances de retarder ou de prévenir des complications graves.
Prévalence
À l’échelle mondiale, sa prévalence est estimée à 1 sur 40 000 à 1 sur 170 000 individus. Cependant, en raison de la complexité des symptômes de la maladie de Fabry et des difficultés liées au diagnostic différentiel, ces chiffres peuvent être sous-estimés.